对bed文件的集合操作是生信分析中的重要组成部分,不过由于集合操作的效果往往五花八门,所以在这里做个最基本的记录。
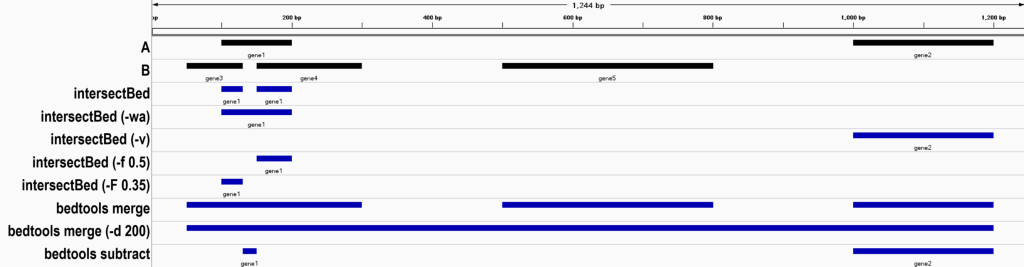
本文的测试环境为 bedtools(v2.31.1),并使用进程替换进行测试,这样就不用创建测试文件了,格式如下:intersectBed -a <(echo -e “chr1\t100\t200”) -b <(echo -e “chr1\t150\t300”)
取交集(intersectBed 等价于 bedtools intersect)
1. 默认操作,输出 A 中与 B 重叠的区间。
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5")
# 结果如下
chr1 100 130 gene1
chr1 150 200 gene12. 保留 A 的完整原始行(即使部分重叠)
参数:-wa (write original A entry)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wa
# 结果如下
chr1 100 200 gene1
chr1 100 200 gene13. 输出 A 中与 B 重叠的区间,并追加的 B 的完整重叠行
参数:-wb
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wb
# 结果如下
chr1 100 130 gene1 chr1 50 130 gene3
chr1 150 200 gene1 chr1 150 300 gene44. 保留 A 的完整原始行以及 B 中完整重叠行
参数:-wa -wb
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wa -wb
# 结果如下
chr1 100 200 gene1 chr1 50 130 gene3
chr1 100 200 gene1 chr1 150 300 gene45. 保留 A 的完整原始行以及 B 中完整重叠行,并输出重叠部分长度
参数:-wo (write overlap)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wo
# 结果如下
chr1 100 200 gene1 chr1 50 130 gene3 30
chr1 100 200 gene1 chr1 150 300 gene4 506. 保留 A 的所有完整原始行以及 B 中完整重叠行,对于在 B 中不存在重叠行的 A,使用“-1”或“.”填充,在最后输出重叠部分长度
参数:-wao (Write All and Overlap)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -wao
# 结果如下
chr1 100 200 gene1 chr1 50 130 gene3 30
chr1 100 200 gene1 chr1 150 300 gene4 50
chr1 1000 1200 gene2 . -1 -1 . 07. 保留 A 的完整原始行,效果与-wa相同,但是去重
参数:-u (write original A entry once if any overlap)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -u
# 结果如下,效果与-wa相同,但是不再有重复行
chr1 100 200 gene18. 仅输出 A 中不与 B 重叠的区间
参数:-v (invert)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -v
# 结果如下
chr1 1000 1200 gene29. 基于重叠比例的过滤
参数:-f -F (minimum overlap fraction)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -f 0.5
# 结果如下,仅输出重叠部分占 A 区间长度 ≥50% 的区间
chr1 150 200 gene1
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -F 0.35
# 结果如下,仅输出重叠部分占 B 区间长度 ≥35% 的区间
chr1 100 130 gene110. 统计 A 中每个区间与 B 的重叠次数(计数模式)
参数:-c (count overlaps)
# 执行命令
intersectBed -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -c
# 结果如下,输出 A 中的片段中
chr1 100 200 gene1 2
chr1 1000 1200 gene2 0取并集
bedtools merge
bedtools merge 用于将重叠或相邻的基因组区间合并为连续的、非重叠的区间。
1. 合并重叠和连续的区间
# 执行命令
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2")
# 结果如下
chr1 50 300
chr1 500 800
chr1 1000 12002. 允许合并的gap长度
参数:-d
# 执行命令
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2") -d 200
# 结果如下,合并间距小于等于200bp的片段为一个独立片段
chr1 50 12003. 合并时保留列信息
参数:-c -o
# 执行命令
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\nchr1\t100\t200\tgene1\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5\nchr1\t1000\t1200\tgene2") -c 4 -o distinct
# 保留了name列信息
chr1 50 300 gene1,gene3,gene4
chr1 500 800 gene5
chr1 1000 1200 gene2-c 和 -o 需要联用。-o 详细如下:
| 类别 | 操作 | 示例 |
|---|---|---|
| 数学统计 | sum,min,max,mean,median,stdev | 10+20 → 30 |
| 极值统计 | absmin(最小绝对值),absmax,mode(众数),antimode(反众数) | -5和3 → 3 (absmin) |
| 列表操作 | collapse(合并值),distinct(去重合并) | “A,B,A”→A,B,A (collapse) |
| 排序列表 | distinct_sort_num(数值升序),distinct_sort_num_desc(降序) | “3,1,2” → 1,2,3 |
| 计数 | count(总数),count_distinct(唯一值数) | “A,B,A” → 3 (count) |
| 提取值 | first,last | “A,B,C” → A (first) |
使用方法:
- 一对一模式:-c 5,4 -o sum,distinct →第5列求和,第4列去重合并。
- 多对一模式:-c 5,6 -o sum → 两列均求和。
- 一对多模式:-c 5 -o sum,mean → 对第5列同时求和和求均值。
4. 链特异性合并
参数:-s
# 执行命令
bedtools merge -i <(echo -e "chr1\t50\t130\tgene3\t.\t+\nchr1\t100\t200\tgene1\t.\t+\nchr1\t150\t300\tgene4\t.\t-\nchr1\t500\t800\tgene5\t.\t+\nchr1\t1000\t1200\tgene2\t.\t+") -s -c 4,6 -o distinct
# 按照链方向合并区间
chr1 50 200 gene1,gene3 +
chr1 150 300 gene4 -
chr1 500 800 gene5 +
chr1 1000 1200 gene2 +bedtools multiinter
可以同时分析多个 BED 文件重叠情况的工具,识别多个文件中共同出现的基因组区域,并统计每个区域在不同文件中的覆盖情况。
1. 基本用法
# 执行命令
bedtools multiinter -i <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5") -names A B
# 结果
chr1 50 100 1 B 0 1
chr1 100 130 2 A,B 1 1
chr1 130 150 1 A 1 0
chr1 150 200 2 A,B 1 1
chr1 200 300 1 B 0 1
chr1 500 800 1 B 0 1
chr1 1000 1200 1 A 1 0取差集(bedtools subtract)
1. 默认操作,从 A 中移除与 B 重叠的区域。
# 执行命令
bedtools subtract -a <(echo -e "chr1\t100\t200\tgene1\nchr1\t1000\t1200\tgene2") -b <(echo -e "chr1\t50\t130\tgene3\nchr1\t150\t300\tgene4\nchr1\t500\t800\tgene5")
# 结果如下
chr1 130 150 gene1
chr1 1000 1200 gene2
发表回复