IGV (Integrative Genomics Viewer) is a powerful and versatile tool for visualizing genomic data, enabling researchers to explore genomic variations effectively. Its graphical interface makes it very user-friendly on Windows systems. However, when batch processing large-scale bioinformatics data, Linux often provides a more suitable environment. Unfortunately, using IGV on Linux can be less straightforward. This article documents the detailed procedures for running and utilizing IGV on Linux systems.
Installation and Setup
Installing IGV
Different versions of IGV require specific versions of Java. Therefore, you should first decide which IGV version you plan to install and then set up the corresponding Java environment.
You can download the IGV installation package from the IGV Download page. Since we will be running IGV on a Linux system, the recommended option is “Command line IGV and igvtools for all platforms (Separate Java 17 required)”. Download the package to your working directory on the server.
After extracting the package, IGV can be launched directly using the igv.sh script included in the installation directory.
# Download the IGV package
wget https://data.broadinstitute.org/igv/projects/downloads/2.18/IGV_2.18.0.zip
# Extract the downloaded archive
unzip IGV_2.18.0.zip
# Run IGV (requires the appropriate Java version to be installed)
cd IGV_2.18.0
igv.shRequirments
IGV requires a specific version of Java to run properly. In addition, generating genomic visualization figures with IGV involves invoking a graphical interface, which depends on an X Window server. On Linux systems, this can be achieved by using Xvfb (X Virtual Frame Buffer) to provide a virtual X server for IGV.
Finally, since the figures exported directly from IGV do not fully meet my requirements, I also installed ImageMagick to perform image cropping and further adjustments.
Creating a Singularity Container
Because IGV requires a specific Java version, managing it directly on a shared Linux environment can be inconvenient. To avoid version conflicts, this guide uses Singularity to create an isolated Linux container dedicated to running IGV. An additional advantage of this approach is portability—simply copy the container to any other Linux system with Singularity installed, and IGV can run immediately without additional configuration.
Singularity is a containerization technology developed by Lawrence Berkeley National Laboratory, designed specifically for large-scale, multi-node HPC and deep learning workloads. It is lightweight, easy to deploy, highly portable, and supports converting Docker images into Singularity images.
Using Singularity is optional—the method described here is simply convenient for managing a clean and reproducible runtime environment. For Singularity installation instructions, refer to “Singularity Installation and Usage Tutorial: A Containerization Guide for High-Performance Computing Environments”.
1. Download a Base Singularity Image
First, we need a Linux base image. You may download an official base image from the Singularity library page, or use the following command to pull a suitable Ubuntu image:
# Pull a base Ubuntu image
[root@comput1 ~/example]$ singularity pull library://library/default/ubuntu
INFO: Downloading library image
28.44 MiB / 28.44 MiB [================================] 100.00% 2.66 MiB/s 10s
WARNING: unable to verify container: ubuntu_latest.sif
WARNING: Skipping container verification
[root@comput1 ~/example]$ ls
ubuntu_latest.sif2. Create a Singularity Sandbox
The downloaded .sif file is read-only. To install software inside it, we must convert it into a writable sandbox directory:
# Convert the sif file into a writable sandbox
[root@comput1 ~/example]$ singularity build --sandbox IGV_env/ ubuntu_latest.sif
INFO: Starting build...
INFO: Creating sandbox directory...
INFO: Build complete: IGV_env/
[root@comput1 ~/example]$ ls
IGV_env ubuntu_latest.sif3. Configure the Environment Inside the Sandbox
Now we can enter the sandbox and install the required dependencies:
# Enter the sandbox in writable mode
[root@comput1 ~/example]$ singularity shell --writable IGV_env/
Singularity>
# Update apt
Singularity> apt-get update
# Install Java 17 (required by the IGV version used in this guide)
Singularity> apt install -y openjdk-17-jdk
# Install Xvfb
Singularity> apt-get install -y xvfb
# Install ImageMagick
Singularity> apt-get install -y imagemagick
# Exit the sandbox
Singularity> exit4. Build a New Read-Only SIF Image
After installing all necessary software, the sandbox can be repackaged into a read-only .sif image, making it more secure and portable:
# Rebuild the sandbox into a read-only sif file
[root@comput1 ~/example]$ singularity build IGV_env.sif IGV_env/
INFO: Starting build...
INFO: Creating SIF file...
INFO: Build complete: IGV_env.sif
[root@comput1 ~/example]$ ls
IGV_env IGV_env.sif ubuntu_latest.sifIf you have successfully created a Singularity container using the steps above, you may now proceed to the section on Generating Genomic Visualization Plots.
If you encounter issues while installing software inside Singularity, or if you choose not to use Singularity at all, refer to the following subsections for installing IGV and its dependencies directly on your Linux system.
Installing Java
1. Installing Java via Package Manager
IGV depends on a specific Java version. You need to install the Java version that matches the IGV release you are using. You can install Java on Linux using the following commands:
apt-get update
# Check available Java versions provided by apt
apt search openjdk | grep -E 'openjdk-.*-jdk/'
# This tutorial uses Java 17
apt install -y openjdk-17-jdk
# Verify Java version
java --version2. Installing Java Manually
# Download the Java package
wget https://download.oracle.com/java/17/latest/jdk-17_linux-x64_bin.tar.gz
# Create an installation directory
mkdir /etc/java-17-openjdk/
# Extract Java to the installation directory
tar -xvf jdk-17_linux-x64_bin.tar.gz -C /etc/java-17-openjdk/
# Add Java to environment variables (temporary)
export JAVA_HOME=/etc/java-17-openjdk/jdk-17.0.12/
export PATH=$PATH:$JAVA_HOME/binInstalling Xvbf
IGV requires a graphical display environment to generate genome visualization plots. However, Linux servers typically do not include a graphical environment by default, and IGV will report errors when attempting to render figures. In such cases, instead of installing a full desktop environment, we can use Xvfb to create a virtual display for IGV.
Xvfb (X Virtual Frame Buffer) is a virtual X Window server that runs graphical applications without physical display hardware or user interaction. It emulates a complete X11 environment—display, keyboard, and mouse—all inside memory. This makes it ideal for automated or headless visualization tasks.
Install Xvfb with:
# Install Xvfb
apt-get install -y Xvfb
# Start Xvfb (Ctrl+C to stop the service)
XvfbIn practical, we typically rely on the command xvfb-run, which starts and stops Xvfb automatically and sets the DISPLAY environment variable correctly. You can start IGV with Xvfb simply by prepending xvfb-run:
# Run IGV with a virtual display
xvfb-run -a --server-args="-screen 0 1280x800x24 -nolisten tcp" igv.sh --batch Batch_scripts_example.txtInstalling ImageMagick
Either due to my limited experience with IGV or potential bugs within IGV itself, the figures generated by IGV were not fully satisfactory for my workflow. Therefore, I also installed ImageMagick to perform additional cropping and refinement on IGV-generated images.
ImageMagick is a powerful image processing suite capable of creating, editing, combining, and converting images. It supports 200+ image formats and provides extensive operations such as resizing, rotation, cropping, watermarking, and filtering. It also supports batch processing and can be used via command line or programmatic interfaces.ImageMagick is open-source and available on Linux, Windows, and macOS.
Install ImageMagick with:
# Install imagemagick
apt-get install -y imagemagick
# Crop an image
# -crop format: WIDTHxHEIGHT+X_OFFSET+Y_OFFSET (origin at top-left corner)
convert example.raw.png -crop 968x485+167+0 example.pngGenerating Genomic Visualization Plots
On Linux, generating genomic visualization plots with IGV is accomplished using the run batch script feature. If you have used IGV on Windows, you may notice that the Batch Script essentially simulates the process of manually creating genome visualizations through the graphical interface. Each user action in the GUI is translated into a corresponding command. These commands are then combined into a batch script file, which instructs IGV on how to generate the genomic visualization plots automatically.
Creating a Batch Script File
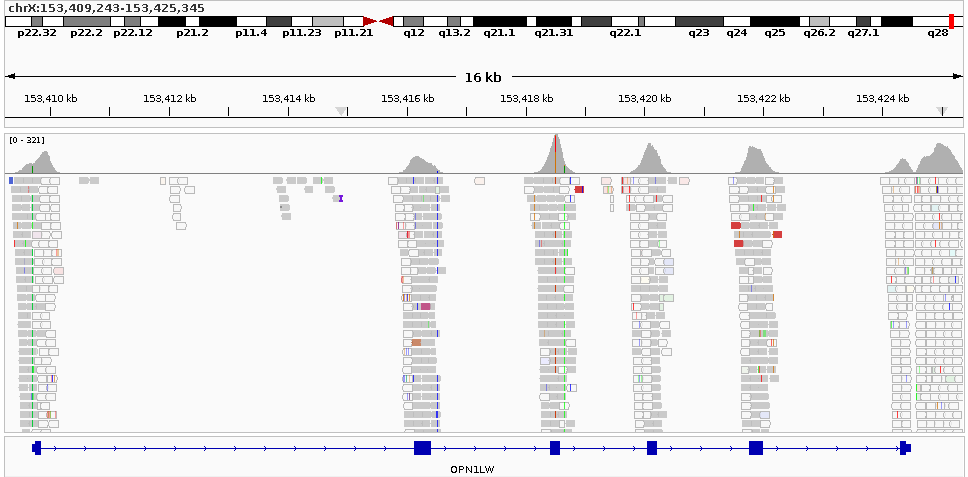
The following batch script can be used to generate a genomic visualization plot for the OPN1LW region:
genome hg19
load ./testSample.deduped.bam name=testSample
goto chrX:153,409,243-153,425,345
sort position
collapse
maxPanelHeight 300
preference NAME_PANEL_WIDTH 0
snapshotDirectory ./mySnapshotDirectory
snapshot testSample.OPN1LW.raw.png
exitBy default, IGV will automatically download and load the hg19 reference genome. However, in most cases, storing the reference genome locally on the server is more stable. You can save the IGV-downloaded genome files to a local server and use a .genome file to instruct IGV how to load the reference genome from the local server.
When IGV automatically downloads the hg19 genome, it provides a .json file with hyperlinks to the required reference genome files. You can use wget to download this JSON file and then retrieve the genome files to your local server using the links provided.
Currently, I have not found a way to generate a .genome file directly on Linux, so this step is performed on Windows. First, install IGV for Windows (Java included) from the IGV Download page. Then transfer the downloaded reference genome files from your local server to the Windows machine. Only the genome sequence files are required; other files such as CytoBand and alias files are optional.
Next, open IGV on Windows and navigate to Genomes -> Create .genome file…. Fill in the genome information and file paths (the Gene field can be left empty; CytoBand and alias files are optional, but the sequence file is required).
Finally, Select the save location for the .genome file.
Tip: It is RECOMMENDED to store the .genome file in the same directory as the reference genome files. This way, the .genome file records only the file names, not the paths information, which facilitates uploading and using the .genome file on Linux.
Once uploaded to Linux, IGV will use the .genome file to locate the reference genome files in THE SAME directory:
hg19/
├─ cytoBand.txt
├─ hg19_alias.tab
├─ hg19.fasta
├─ hg19.fasta.fai
├─ hg19.genome
└─ ncbiRefSeq.txt.gzTo load the local .genome file, the batch script must be modified slightly. In particular, the ncbiRefSeq.txt.gz file should be loaded separately, as it provides the gene annotations displayed at the bottom of the genomic plot. Since the required Gene file format for generating .genome files is unclear for me, I manually added the gene file as follows:
genome ./genome/hg19/hg19.genome
load ./genome/hg19/ncbiRefSeq.txt.gz name=Gene
load ./testSample.deduped.bam name=testSample
goto chrX:153,409,243-153,425,345
sort position
collapse
maxPanelHeight 300
preference NAME_PANEL_WIDTH 0
snapshotDirectory ./mySnapshotDirectory
snapshot testSample.OPN1LW.raw.png
exitRunning IGV
You can create a shell script (run.sh) to facilitate running IGV inside the previously prepared Singularity container:
# If Java was installed manually, uncomment the following lines to set temporary environment variables
# export JAVA_HOME=/etc/java-17-openjdk/jdk-17.0.12/
# export PATH=$PATH:$JAVA_HOME/bin
# Run IGV using Xvfb to generate the visualization plot.
# IGV executes commands from the batch file to produce the genomic visualization.
xvfb-run -a --server-args="-screen 0 2560x720x24 -nolisten tcp" /root/shenh/singularity/IGV/IGV_2.18.0/igv.sh --batch ./Batch_script.txt
# Crop the IGV-generated genomic plot
convert ./mySnapshotDirectory/testSample.OPN1LW.raw.png -crop 968x485+167+0 ./mySnapshotDirectory/testSample.OPN1LW.pngIn principle, the preference NAME_PANEL_WIDTH 0 command in the batch script should remove the Name Panel on the left side of the visualization. However, for some reason, it does not take effect. Therefore, I used ImageMagick to crop the left panel manually.
Run the following commands to generate the genomic visualization plots:
# 对于使用了Singularity的用户,命令如下
singularity exec IGV_env.sif sh run.sh
# 对于未使用Singularity的用户,命令如下
sh run.sh
Leave a Reply